
Human Enteroviruses and Chronic Infectious Disease
Steven Tracy and Nora M. Chapman
(from the Journal of IiME Volume 3 Issue 1Professor Steven Tracy Ph.D.
Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha NE 68198-6495; stracy@unmc.edu; 402-559-7747
Ph.D., 1979, Department of Biology, University of California, San Diego
Primary research interest: Molecular biology and pathogenesis of the group B coxsackieviruses

ABSTRACT
Most of what is known about human enteroviruses (HEV) has been derived from the study of the polioviruses, the HEV responsible for poliomyelitis. The HEV are generally not thought to persist for long periods in the host: an acute, sometimes nasty, infection is rapidly eradicated by the host's serotype-specific adaptive immune response.
Our discovery that the commonly encountered HEV, the group B coxsackieviruses (CVB), can naturally delete sequence from the 5' end of the RNA genome and that this deletional mechanism results in long-term viral persistence, in the face of the adaptive immune response, has substantially altered this view.
This previously unknown and unsuspected aspect of enterovirus replication provides an explanation for previous reports of enteroviral RNA detected in diseased tissue in the apparent absence of infectious virus particles.
Introduction
The enteroviruses are an incredibly diverse and large genus in the family Picornaviridae. Within the enteroviruses, human enteroviruses (HEV; those which infect humans as opposed to other species) number at least 100 known serotypes, with more known to exist but which have just not been characterized to date. Serotype defines the virus: it is how the immune system recognizes the complex aggregation of proteins which makes up the virus particle or virion.
Thus, infection with one HEV serotype induces immunity that protects against disease which that HEV serotype might inflict upon one were the virus is encountered again, but this protective immunity does not extend to other serotypes. This is why the poliovirus vaccines work to protect from poliomyelitis: any subsequent infection with poliovirus is quickly suppressed by the pre-existing anti-poliovirus immunity and the virus infection eradicated. But of course, anti-poliovirus protective immunity does not protect from being infected with a different HEV. Human enterovirus virions are small at 29nm in diameter; said another way, this means about 345,000 viruses would need to be lined up to equal one centimeter.
The virus particle consists of an ordered array of 4 capsid proteins that forms an icosahedral structure of incredible beauty.
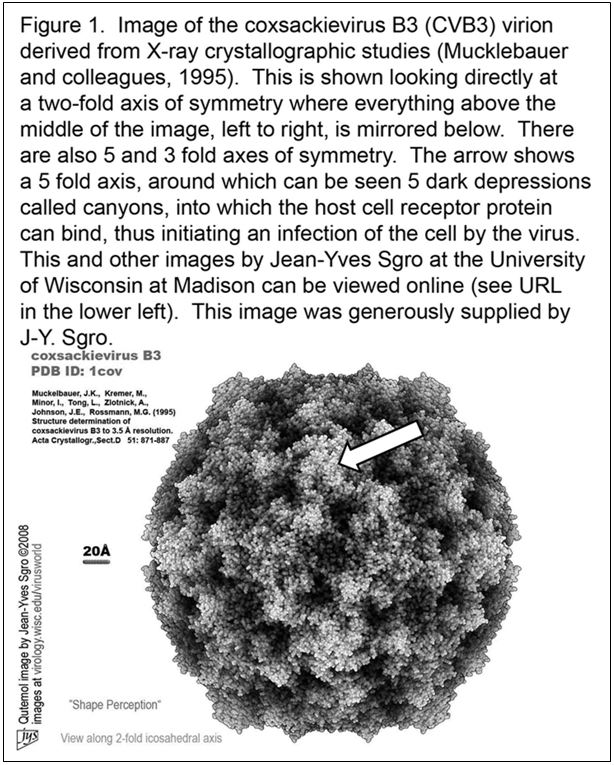
In Figure 1, the reader can see what the coxsackievirus B3 (CVB3) virion would look like were one able to actually see it.
This structure was solved with a technique called X-ray crystallography (23) in which X-rays, which are directed through crystals of virus and bent in specific ways, are then interpreted by computer analysis to provide a virus structure.
Numerous other images of related enteroviruses can be found by visiting the website of the Institute for Molecular Virology at the University of Wisconsin in Madison WI, USA.
The HEV cause a plethora of different human diseases and syndromes, the most important of which are poliomyelitis [now largely, although not entirely, eradicated in the world through use of the vaccines (16)], meningitis and encephalitis, myocarditis, pancreatitis, myositis, and type 1 (insulin dependent) diabetes (T1D) (31).
Recent work has also indicated a possible role for HEV in the poorly understood etiology of chronic fatigue syndrome (CFS) (9, 11). Polio, T1D and CFS are noteworthy, in that these diseases are chronic and in the case of polio and T1D, fatal without treatment. Certainly, fatal cases of the other HEV diseases occur as well (28) but as a rule, HEV diseases are deemed to be acute illnesses of relatively low clinical importance because they are so common. Nonetheless, 5-10 million cases of symptomatic HEV infections occur annually in the US alone (27). Group B coxsackievirus infection of the heart
Within only a few years after their discovery in the late 1940s, the group B coxsackieviruses (CVB) were shown to be involved in inflammatory heart disease or myocarditis (6,12, 32). Because of the ease with which the CVB replicated in mice, an experimental model to study myocarditis was soon available and has been exploited for numerous studies over the years. Once myocardial biopsy techniques became widespread as a clinical assay for the presence of myocarditis (29), researchers became interested in determining how often HEV was associated with myocarditis. These studies have demonstrated that about 15-20% of adult myocarditis cases can be associated with an HEV infection (2). This was carried out in most cases by isolating RNA from very small biopsy samples of the human heart, then analyzing the RNA for the presence of HEV RNA using a variety of techniques.
Interestingly, in those adult cases of myocarditis in which the presence of virus was shown by detecting the viral RNA, rarely can an infectious virus be isolated. This is confusing: how can one detect viral RNA and not detect the virus? And indeed, this was a conundrum for many years. Most HEV cause cells in culture to die; this outcome, termed cytopathic effect or cpe, is the result of the virus infecting the cells in culture and killing them in the process of producing the next virus generation. Failure to observe cpe upon inoculating cell cultures with homogenized heart samples, was taken to be evidence that virus qua virus was not present. This goes back to the foregoing discussion, in which the general view of HEV is as a virus that rapidly causes cell lysis (acute disease). As opposed to adult samples, samples of pediatric myocarditic heart tissue generally shows both the presence of cytopathic virus when placed in culture as well as the presence of viral RNA when molecular assays are carried out.
We considered the possibility that HEV infections of adult human hearts might generate a population of viruses that have been characterized and termed defective interfering (DI) viruses (10,13). Although DI HEV had never been demonstrated in humans or animals, they had been shown to exist in experimental cell culture and thus, the possibility that they might also exist in nature could not be excluded. Defective interfering HEV are viral RNA genomes that have deleted variable parts of the sequence that encode the capsid (coat) proteins of the virus (see Figure 2). This implies such viruses could never be successful, as they could never produce an intact virion. However, DI HEV exist in a dynamic equilibrium with so-called wild type HEV which do produce normal capsids. Therefore, the DI viruses are parasites upon the wild type population of HEV, using their capsids to package the mutated RNA for movement to the next cell. Using a mouse model of CVB3-induced myocarditis, in which viral RNA was detectable in the heart tissue for many days after cytopathic virus was no longer detectable in cell culture, we searched for evidence of DI forms of CVB3 RNA but were repeatedly unsuccessful.
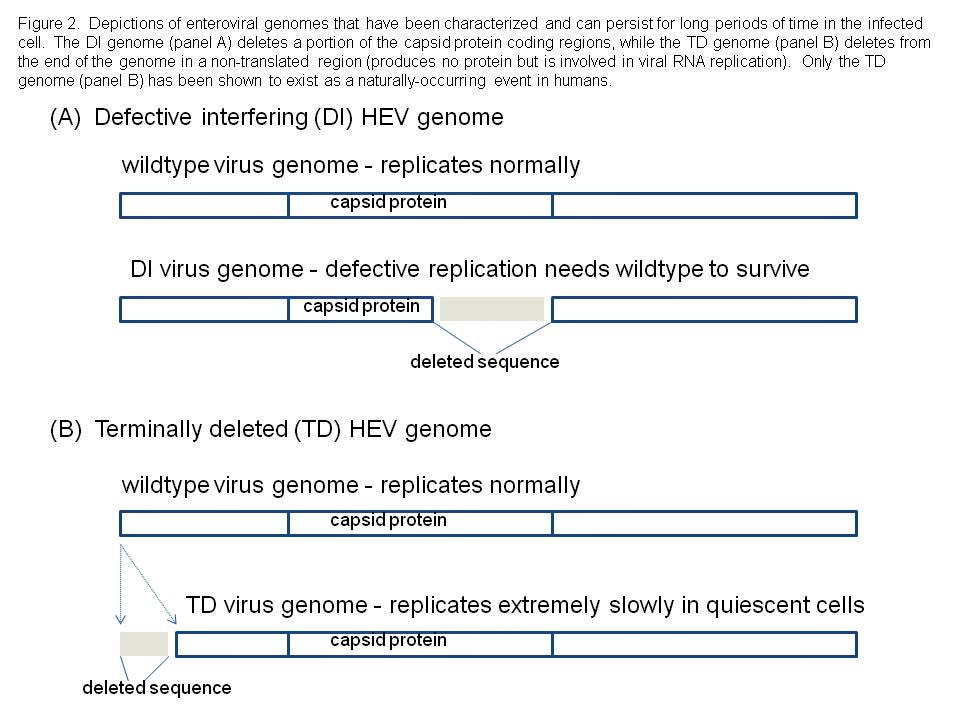
We decided to examine the entire viral RNA genome that was present in these mouse heart samples, asking the basic question: are there deletions anywhere else that might explain this odd phenomenon? When this was done, we discovered that one of the ends of the single strand of RNA that makes up the viral genome, was missing: these viral genomes were then called 'terminally deleted' or TD (8, 18, 19). What makes this discovery fascinating to virologists, is that the sequence which the virus naturally deletes, was hitherto thought to be absolutely essential for virus replication. Our results, however, showed that while this sequence was very important for efficient CVB replication, it could nonetheless be done away with, and yet have the virus survive.
The cost of this survival is, however, extremely slow replication. A further cost is that this survival can occur only in cell populations that do not divide anymore or divide very infrequently as in muscle tissue. It is this reason why we were able to find these novel virus populations in heart muscle of experimentally inoculated mice (19) and later, in human heart (8). There are some cell cultures that do not divide continually but stop dividing when the cells contact each other; only in such cultures can CVB-TD populations occur (18). This is very different than current cell culture models for enterovirus infections in which most aspects of the viral biology are examined in immortal continuously replicating cells. While this is a good model for the short term infection of the gastrointestinal tract in which the virus infects, rapidly produces progreny virus and is excreted to infect another individual before the immune response can curtail infection, much of the pathology associated with enterovirus infections is in other differentiated tissues in which relatively little cell turnover may occur. In these non-replicating (or only intermittently replicating) cells, the wild type virus is at a disadvantage because these cells lack key cytoplasmic factors essential for rapid replication. However, the low level replication of the TD viruses is favored in these cells and as the intracellular portion of the virus replication cycle is much longer, it is relatively hidden to the immune system. Much of our current research focus is upon the mechanism of selection of the TD populations in such cell cultures or tissues.
These results provided an answer to the conundrum of failing to find cytopathic virus in myocarditic heart samples despite the ability to find viral RNA. Indeed, virus (in TD form) does exist in such samples but because the TD populations replicate so slowly and produce so little virus, they are difficult to detect. However, because the defect is not in a part of the viral genome that makes viral proteins, they do make all the viral proteins and even virus particles. But what does the finding of TD genomes mean for HEV disease?
Chronic disease associated with HEV infections
The previous discussion has demonstrated that HEV can persist for longer periods of time in the immunologically-normal host than ever had been suspected but that this came at a price to the virus: very slow replication and relatively very few infectious particles produced.
In cases of chronic inflammatory heart disease (chronic myocarditis) or what is often thought to be a sequela of this condition, called dilated cardiomyopathy (DCM), HEV RNA has been detected in the apparent absence of cytopathic virus (4, 7, 21). We can now say with some certainty that in such cases, HEV-TD populations were present. In DCM, the heart is failing due to damage to the cardiomyocytes (muscle cells of the heart). Others have shown that a HEV enzyme that works on and reduces proteins, called a protease, can damage an important cardiomyocyte protein called dystrophin (3) and that in a mouse model of this disease (33), dystrophin is cleaved despite the inability of the virus to produce infectious particles and this leads to DCM in the mouse, a scenario that is closely similar to the low level production of virus particles by TD virus populations. Therefore, in this chronic HEV-induced disease, the long-term persistence of a slowly replicating HEV can lead to cell damage, due to the virus' own enzymes it uses to replicate, and this damage eventually impacts the function of the organ (the heart) itself.
Another chronic disease that is closely linked to HEV infections is type 1 (insulin dependent) diabetes (or T1D) (15, 30). Type 1 diabetes occurs due to an inability to control glucose metabolism which is an outcome from the loss of insulin-producing beta cells in the pancreatic Islets of Langerhans (17). Although some cases are thought to be due only to a specific expression of individual genetic traits, most T1D cases cannot be so easily explained and therefore, environmental factors (like infections) have been sought to explain how T1D is initiated (1, 20). One environmental factor that is high on any list, are the HEV: many clinical observations and experimental studies implicate HEV as agents that can and do trigger T1D onset in humans (15, 30). While CVB have been associated with T1D cases, other non-CVB HEV have also been implicated in T1D onset, further adding to the evidence for a role of HEV in T1D onset. At present, it is unclear whether the HEV involved persists in the host after the initial infection that sets the disease in motion, or whether it is more of a classical HEV acute infection, one that is rapidly cleared by the immune response. What is very clear, however, is that CVB can rapidly trigger T1D onset in a T1D-prone mouse called the NOD mouse if the mouse is already prediabetic from its own autoimmune attack on its pancreatic islets (14). This means that under certain circumstances (when one has autoimmune insulitis present, and one is infected with an HEV against which one has no pre-existing protective immunity, and it is the correct HEV at the right dose), T1D in humans could likely be initiated by an HEV infection. Using the rapid onset model in mice to make inferences for humans, we would predict an HEV infection that rapidly kills enough beta cells will initiate T1D. However, the virus infection might not accomplish this: the virus might instead kill insufficient numbers of beta cells for T1D to ensue. What then? This is where the autoimmune (in which one's immune system attacks oneself) aspect of T1D sets this disease apart from the previously discussed chronic heart disease. In T1D, enough insulin-producing beta cells must be destroyed in order for T1D to occur: this can happen by autoimmune processes, by virus attack, or both occurring together. We are currently assessing whether long-term persistence of HEV is a factor in both the NOD mouse model of T1D and in human beings.
Chronic fatigue syndrome and the link to HEV infection
In a paper that received much notice, Chia and Chia showed that HEV RNA and protein were detectable in the great majority of stomach biopsy tissue samples from patients diagnosed with chronic fatigue syndrome (CFS) but only in few biopsy samples from control patients without the disease (9). This report has suggested that a commonly circulating human virus group might be a primary etiologic agent involved in CFS, a disease that is marked by a difficult diagnosis and a near complete lack of understanding about what agent(s)/mechanism(s) trigger the disease.
As we have seen from the previous discussion, HEV may be able to initiate a disease process just from an acute infection, which is then resolved, or from a continuing infection as well, involving a persistent virus population. Persistence is, however, a relative term. In an immunologically-normal individual, i.e., one who is able to mount normal vigorous immune responses against infectious agents, an HEV infection may be able to persist via a TD genome mechanism for some weeks, perhaps months, but eventually will be eradicated by the immune response. Thus, such infections are temporary (unlike herpesviruses or HIV). Although HEV are common viruses, the very high positive correlation with the CFS stomach samples was surprising. In other known associations of HEV with human diseases, such high correlations have not been observed. Poliovirus caused paralytic disease during epidemics (22) but only about 1 out of every 100-200 infections involved life-threatening paralysis. The HEV, thought to be primarily CVB, which have been closely linked to causing myocarditis, have been detected in about 15-20% of samples in a variety of studies (21).
To explain the high number of positive stomach samples with CFS, one must consider the possibility of a continuing process in CFS patients of new infections with different HEV serotypes and/or a significant number of persistent infections in the stomach. The difficulty these workers had in culturing the viruses from stomach tissue, would be consistent with either HEV strains that do not replicate well in standard tissue culture systems and/or the viruses exist in a form that is difficult to detect. While most HEV do replicate in certain cell cultures, others require the use of suckling mice (27). It is interesting to consider that a HEV-TD population would fit such a description of a 'difficult to culture virus".
At present, these findings are highly intriguing but need also to be considered with the proverbial 'grain of salt'. It is of the highest and immediate importance to identify the HEV in these stomach biopsies in order to verify and move forward the theory that HEV are involved in the CFS etiology. Are they a specific serotype of HEV or are they many different types?
This can be determined without culturing the viruses, by amplification of specific genome regions and determination of the RNA sequences (5, 24-26). Once established which specific HEV are present, relevant model systems might be developed using cell cultures and possibly mice, to study the CFS disease onset process.
Summary
Basic research into the biology of the HEV has lead to truly fantastic discoveries which in turn, have lead to 'bench to bedside' advances. Understanding how to culture animal cells in the laboratory, made possible propagation of the agent that causes polio and subsequent studies in primates, work that in turn lead to the development of the successful poliovirus vaccines from which we have all profited. A search for other viral agents that induce polio-like disease lead to the discovery of the coxsackieviruses and indeed many other HEV.
The development of a mouse to study retinal disease produced the NOD mouse, a highly useful mouse strain that is regularly used in studies of T1D. We know that serendipity and planning go hand in hand and often in the laboratory, are hard to tell where one begins and the other leaves off.
As scientists who focus on all things enteroviral, we are especially interested in the findings of Chia and Chia that suggest the potential for an HEV link in CFS etiology.
Our finding that a deletion in the terminal portion of the CVB genome, was the mechanism by which HEV can persist, could not have been predicted based on what was known. This finding has opened a completely new and quite unsuspected chapter in the very well thumbed volume on enterovirus biology.
This was elegantly enunciated by Louis Pasteur in his famous quotation: "Where observation is concerned, chance favors the prepared mind." Without the existence of basic science, most of what we know take for granted in medicine would not exist.
It is of the greatest importance to keep in mind the goal toward which one works in science, but it is also of equal importance to simply explore and define the 'new' while keeping that mind well prepared for finding new treasures.
It is only through such efforts that we believe the etiology of CFS will be finally illuminated.
Acknowledgements.
Much of our work has been variously supported by the National Institutes of Health, the American Heart Association, the Juvenile Diabetes Research Foundation, and the American Diabetes Association.
We also sincerely thank a group of concerned individuals who have lost family due to enteroviral heart disease, for their generous support.
References
1 ADDIN EN.REFLIST Akerblom, H. K., O. Vaarala, H. Hyoty, J. Ilonen, and M. Knip. 2002. Environmental factors in the etiology of type 1 diabetes. Am J Med Genet 115:18-29.
2 Baboonian, C., M. J. Davies, J. Booth, and W. McKenna. 1997. Coxsackie B viruses and heart disease. Curr Top Microbiol Immunol 223:31-52
3 Badorff, C., N. Berkely, S. Mehrotra, J. W. Talhouk, R. E. Rhoads, and K. U. Knowlton. 2000. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin-based substrate analogue. J Biol Chem 275:11191-7.
4 Bowles, N. E., P. J. Richardson, E. G. Olsen, and L. C. Archard. 1986. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1:1120-1123.
5 Brown, B., M. S. Oberste, K. Maher, and M. Pallansch. 2003. Complete genomic sequencing shows that polioviruses and members of human enterovirus species C are closely related in the noncapsid coding region. J Virol 77:8973-8984.
6 Chapman, N., A. Ramsingh, and S. Tracy. 1997. Genetics of coxsackievirus virulence. Curr Top Microbiol Immunol 223:227-258.
7 Chapman, N. M., and K.-S. Kim. 2008. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Topics Microbiol Immunol 323:275-292.
8 Chapman, N. M., K. S. Kim, K. M. Drescher, K. Oka, and S. Tracy. 2008. 5' terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology.
9 Chia, J. K., and A. Y. Chia. 2008. Chronic fatigue syndrome is associated with chronic enterovirus infection of the stomach. J Clin Pathol 61:43-8.
10 Cole, C. N., D. Smoler, E. Wimmer, and D. Baltimore. 1971. Defective interfering particles of poliovirus. I. Isolation and physical properties. J Virol 7:478-485.
11 Cunningham, L., N. E. Bowles, R. J. Lane, V. Dubowitz, and L. C. Archard. 1990. Persistence of enteroviral RNA in chronic fatigue syndrome is associated with the abnormal production of equal amounts of positive and negative strands of enteroviral RNA. J Gen Virol 71:1399-1402.
12 Dalldorf, G. 1955. The coxsackie viruses. Annu Rev Microbiol 9:277-296.
13 Dimmock, N. J. 1991. The biological significance of defective interfering viruses. Rev Med Virol 1:165-176.
14 Drescher, K. M., K. Kono, S. Bopegamage, S. D. Carson, and S. Tracy. 2004. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: Insulitis determines susceptibility of pancreatic islets to virus infection. Virology 329:381-394.
15 Drescher, K. M., and S. Tracy. 2008. The CVB and etiology of type 1 diabetes. Curr Topics Microbiol Immunol 323:259-274.
16 Dutta, F. 2008. Epidemiology of poliomyelitis - options and update. Vaccines 26:5767-5773.
17 Haller, M. J., M. A. Atkinson, and D. Schatz. 2005. Type 1 diabetes mellitus: etiology, presentation, and management. Pediatr Clin North Am 52:1553-78.
18 Kim, K. S., N. M. Chapman, and S. Tracy. 2008. Replication of coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol 82:2033-7.
19 Kim, K. S., S. Tracy, W. Tapprich, J. Bailey, C. K. Lee, K. Kim, W. H. Barry, and N. M. Chapman. 2005. 5'-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 79:7024-41.
20 Knip, M., and H. K. Akerblom. 1999. Environmental factors in the pathogenesis of type 1 diabetes mellitus. Exp Clin Endocrinol Diabetes 107 Suppl 3:S93-S100.
21 Martino, T. A., P. Liu, M. Petric, and M. J. Sole. 1995. Enteroviral myocarditis and dilated cardiomyopathy: a review of clinical and experimental studies., p. 291-352. In R. HA (ed.), Human Enterovirus Infections. ASM Press, Washington DC.
22 Minor, P. D. 2003. Polio vaccines and the cessation of vaccination. Exp Rev Vaccines 2:99-104.
23 Mucklebaur, J. K., and M. G. Rossmann. 1997. The structure of coxsackievirus B3. Curr Top Microbiol Immunol 223:191-208.
24 Oberste, M. S., K. Maher, M. R. Flemister, G. Marchetti, D. R. Kilpatrick, and M. A. Pallansch. 2000. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J Clin Microbiol 38:1170-1174.
25 Oberste, M. S., K. Maher, D. Kilpatrick, M. Flimister, B. Brown, and M. A. Pallansch. 1999. Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol 37:1288-1293.
26 Oberste, M. S., K. Maher, A. J. Williams, N. Dybdahl-Sissoko, B. A. Brown, M. S. Gookin, S. Penaranda, N. Mishrik, M. Uddin, and M. A. Pallansch. 2006. Species-specific RT-PCR amplification of human enteroviruses: a tool for rapid species identification of uncharacterized enteroviruses. J Gen Virol 87:119-28.
27 Pallansch, M. A., and R. Roos. 2007. Enteroviruses: Polioviruses, coxsackieviruses, echoviruses and newer enteroviruses, p. 839-893. In D. M. Knipe (ed.), Fields Virology, vol. 1. Wolters Kluwer, Philadelphia.
28 Romero, J. T. 2008. Pediatric group B coxsackievirus infections. Curr Topics Microbiol Immunol 323:223-239.
29 Shanes, J., J. Ghali, M. Billingham, V. Ferrans, J. Fenoglio, W. Edwards, C. Tsai, J. Saffitz, J. Isner, S. Furner, and e. al. 1987. Interobserver variability in the pathologic interpretation of endomyocardial biopsy results. Circulation 75:401-405.
30 Tracy, S., and K. M. Drescher. 2007. Coxsackievirus infections and NOD mice: Relevant models of protection from, and induction of, type 1 diabetes. Ann N Y Acad Sci.
31 Tracy, S., M. S. Oberste, and K. M. Drescher. 2008. Group B Coxsackieviruses. Curr Topics Microbiol Immunol 323.
32 Van Creveld, S., and H. De Jager. 1956. Myocarditis in newborns, caused by coxsackie virus. Ann Pediat 187:100-112.
33 Wessely, R., K. Klingel, L. F. Santana, N. Dalton, M. Hongo, W. Jonathan Lederer, R. Kandolf, and K. U. Knowlton. 1998. Transgenic expression of replication-restricted enteroviral genomes in heart muscle induces defective excitation-contraction coupling and dilated cardiomyopathy. J Clin Invest 102:1444-53.

Further Reading:
Dr John Chia/Dr Andrew Chia - click here
Enterovirus Foundation - click here
